Peptides are gaining recognition as effective middle-sized therapeutic agents in the medical field. Unlike small-molecule drugs, peptides have the ability to target complex biological processes with greater precision. Additionally, peptides are generally less complex and more cost-effective than larger biological drugs such as antibodies. The development of peptides as therapeutic agents has led to over 100 FDA-approved peptide drugs entering the market since the creation of the first peptide hormone, insulin, in 1923. These peptide drugs have been utilized in the treatment of a wide range of diseases, including cancer, cardiovascular issues, and metabolic diseases.
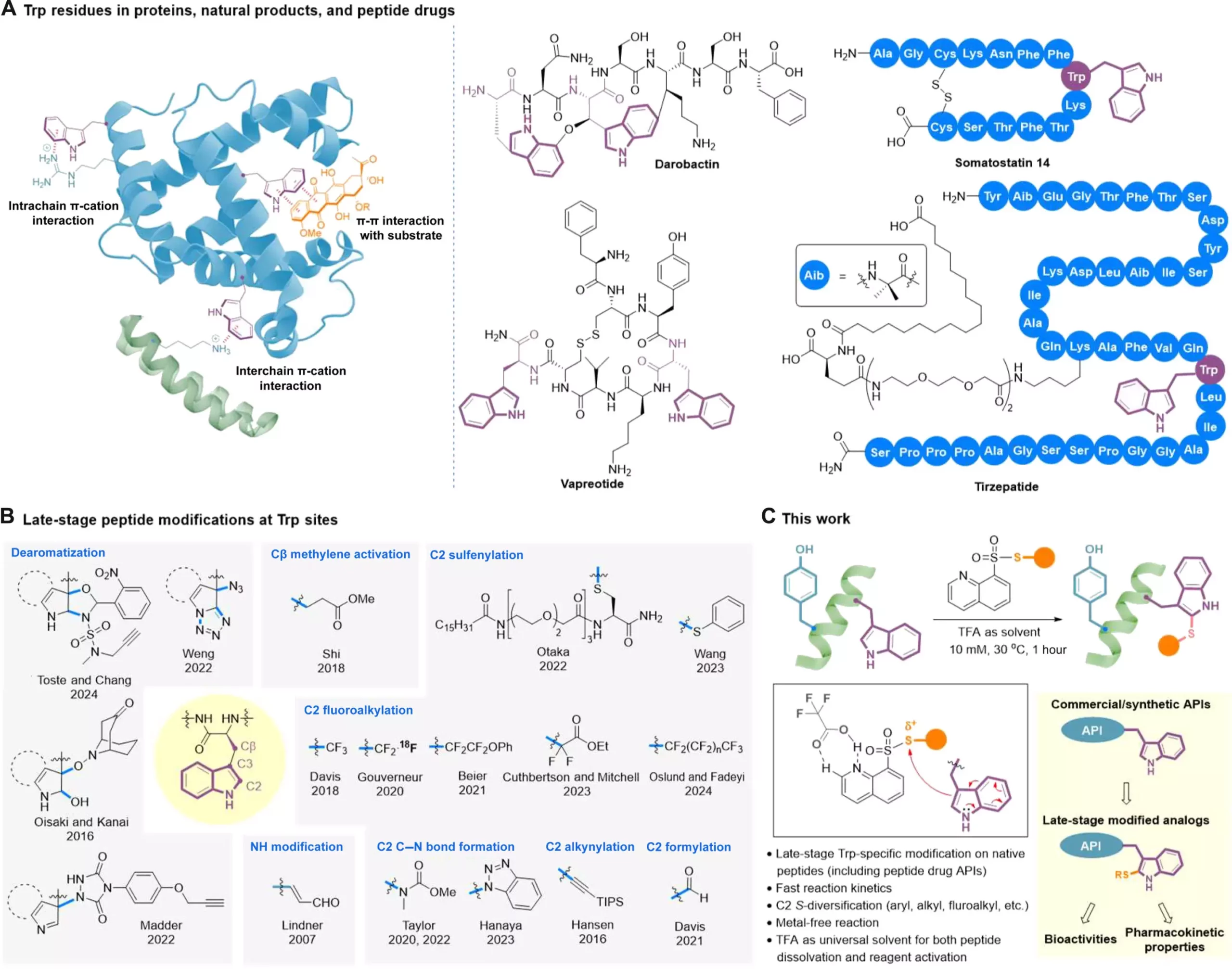
It is noteworthy that around 40 of these FDA-approved peptide drugs contain at least one tryptophan (Trp) residue, which is a crucial amino acid. Modifying Trp residues within peptide molecules can significantly impact drug-target interactions, as well as enhance drug stability, bioavailability, and pharmacokinetic properties. However, the process of making modifications to these functionally dense molecules requires a high level of selectivity, including chemoselectivity, regioselectivity, and stereoselectivity. Additionally, peptides possess nucleophilic functionalities that make them sensitive to redox conditions, further complicating the modification process.
One of the major challenges in the development of peptide drugs is the limited solvents available for dissolving unprotected peptides. This poses an obstacle when attempting to conduct site-specific late-stage peptide modifications. Despite these challenges, a team led by Professor Xuechen Li from the Department of Chemistry at The University of Hong Kong has made significant progress in this area. They have devised a clickable tryptophan modification strategy that enables the straightforward modification of specific regions within a peptide molecule, even during the later stages of drug development.
The technique developed by Professor Li’s team involves a late-stage catalyst-free C2-sulfenylation reaction utilizing S-modified quinoline-containing thiosulfonate reagents. Through this method, a wide range of functional groups can be efficiently installed onto tryptophan (Trp) residues within native peptide structures. These functional groups include trifluoromethylthio, difluoromethylthio, (ethoxycarbonyl) difluoromethylthio, alkylthio, and arylthiol. Trifluoroacetic acid (TFA) plays a crucial role in this transformation by acting as the optimal solvent and facilitating the activation of reagents through hydrogen bond interaction.
The method developed by Professor Li’s team has been successfully applied to the late-stage modification of several on-market peptide drugs, such as somatostatin, octreotide, lanreotide, setmelanotide, daptomycin, and semaglutide. Additionally, it has been utilized in the modification of the bioactive glycopeptide hAdn-WM6877. This demonstrates the versatility and applicability of the method in diversifying peptide-based active pharmaceutical ingredients. The team has observed improvements in the bioactivity and serum stability of modified melittin analogs, highlighting the method’s potential in drug development.
The Future of Peptide Modification
Considering the widespread presence of Trp in RiPP natural products and drug leads from phage display and mRNA display, this modification method holds promise for natural product late-stage diversification. It could serve as a valuable tool in generating molecular libraries and functional probes. The single-step clickable late-stage Trp modification method has the potential to become a robust platform for producing structural analogs in a cost-effective manner. It is poised to meet the demand for optimizing drug activities and pharmacokinetic properties, making it a valuable asset for medicinal chemists, peptide chemists, and chemical biologists alike.
Leave a Reply