Molecules, the fundamental building blocks of matter, consist of intricately bonded atoms, each contributing to a web of interactions dictated by their quantum mechanical nature. At their core lies a nucleus laden with positive charges, while the chaotic dance of negatively charged electrons occurs in the surrounding space. This dance is governed by the laws of quantum mechanics, specifically articulated in the Schrödinger equation, which determines the possible energy levels and states of a quantum system. However, deciphering this equation for molecules, especially those with substantial atomic counts, is no simple endeavor. The computational load associated with simulating these interactions demands advanced tools beyond the capabilities of traditional methods.
The Groundbreaking Leap in Machine Learning
Researchers at the Berlin Institute for the Foundations of Learning and Data (BIFOLD) in collaboration with Google DeepMind have ventured into this challenging realm, developing a novel machine learning algorithm that enhances molecular dynamics simulations. Their advancements, published in Nature Communications, promise to revolutionize the study of molecular interactions, enabling scientists to explore the nuances of complex chemical systems over extensive time periods with remarkable efficiency.
Traditionally, molecular dynamics simulations rely heavily on resolving the intricate interactions delineated by the Schrödinger equation. For molecules comprising dozens of atoms or more, the computational burden can extend into days, even on high-performance computing platforms. This is compounded further when simulations require solving the equation countless times for long-term dynamics. The consequential limitations of time and resource constraints have long stifled innovations in chemical research.
A Paradigm Shift with Reduced Computational Costs
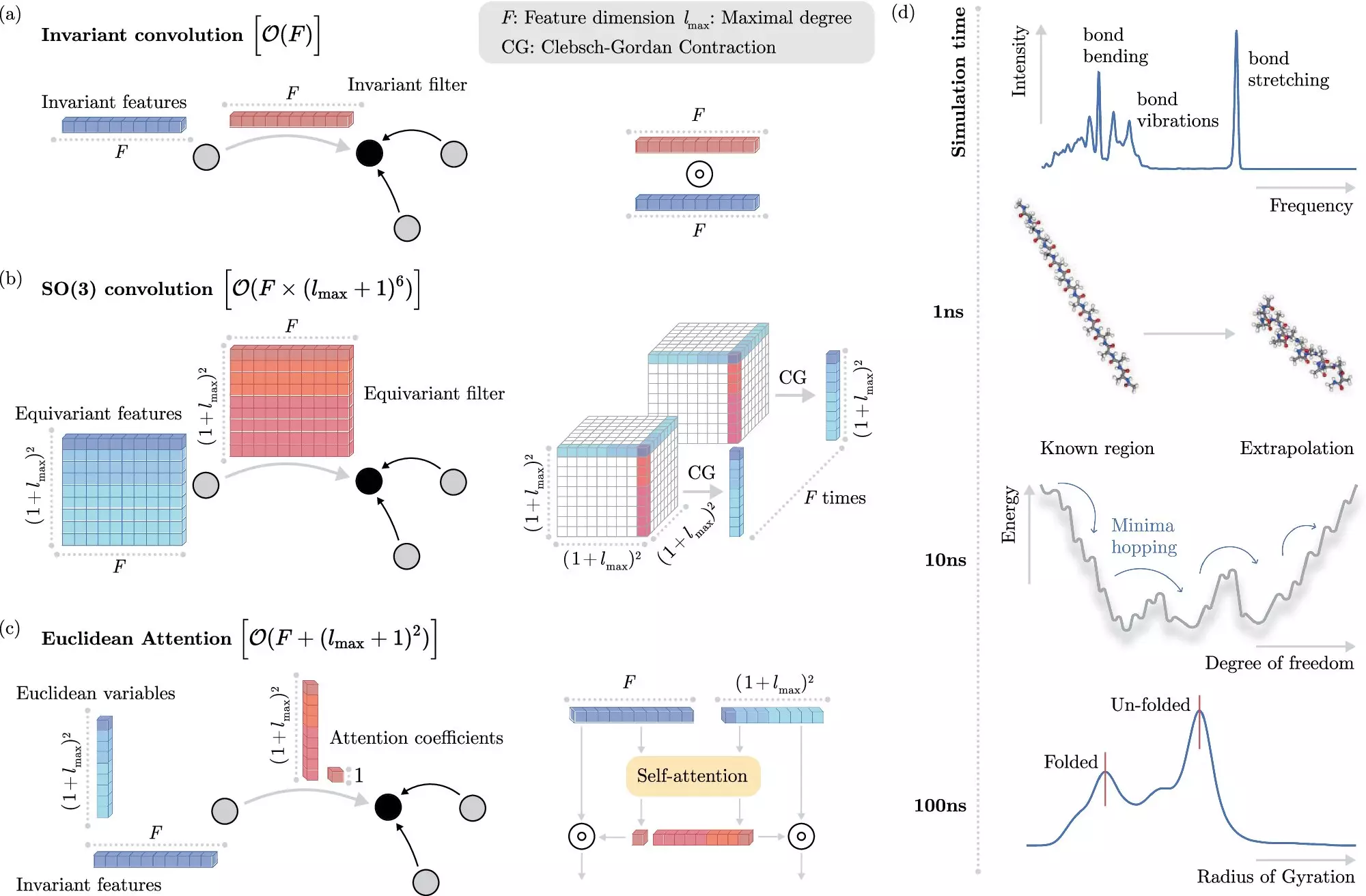
The BIFOLD researchers’ machine learning-based approach shifts the focus away from the explicit resolution of equations towards predictive modeling of electronic interactions. By training algorithms to anticipate the outcomes of these interactions, researchers can sidestep the computational bottleneck posed by direct calculations. This transformative capability not only accelerates the speed of simulations but also brings them to scales previously deemed unattainable, enabling precise modeling of intricate processes such as protein folding and molecular binding.
In this innovative framework, the BIFOLD team addressed the fundamental challenge of incorporating physical invariances into their machine learning models. Invariances allow certain properties of molecular systems to remain constant, streamlining the learning process. However, prior techniques often rendered the addition of these invariances computationally intensive, limiting overall efficiency. The BIFOLD team’s solution elegantly decouples these invariances from the core operations within the model, reserving complex computations for truly significant physical interactions. This shift has led to dramatic reductions in required computation time, enabling simulations that previously took months to complete to now be executed in mere days on a single computing node.
Applications Beyond the Laboratory
As this advanced methodology matures, the implications for drug development emerge as particularly significant. Traditional approaches in pharmaceutical research often necessitate exhaustive empirical experimentation, which can be both time-consuming and costly. The capability to accurately simulate molecular interactions within the human body opens a pathway towards the development of new therapeutic agents without the need for extensive trial-and-error lab work. Such processes not only expedite drug discovery but also contribute to environmental sustainability by reducing the quantity of physical experiments required.
One remarkable application tackled by the researchers involved identifying stable configurations of docosahexaenoic acid (DHA), an essential fatty acid integral to human brain structure. This task, which would typically necessitate the analysis of tens of thousands of molecular candidates using conventional quantum mechanics, was accomplished efficiently through their machine learning framework, illustrating the technique’s transformative potential in real-world scenarios.
The Road Ahead: Challenges and Future Directions
As exciting as these advancements are, challenges still loom on the horizon. The next generation of algorithms must grapple with simulating even larger systems with accurate descriptions of complex, long-range physical interactions. The intersection of machine learning with physical reality in computational chemistry is not just a scientific endeavor; it’s a multidisciplinary approach that promises to redefine how we understand and manipulate matter at the atomic level.
Prominent voices in the field, including Prof. Dr. Klaus-Robert Müller, co-director at BIFOLD and a principal scientist at Google DeepMind, underscore the importance of continuing this exploratory research. By marrying cutting-edge machine learning techniques with the principles of physical sciences, researchers are poised to make substantial strides toward solving long-standing questions in computational chemistry. The journey to harnessing these powerful innovations is only just beginning, marking an exciting era for science and technology as they converge to push the boundaries of what’s possible.
Leave a Reply